 |
これらの違い | 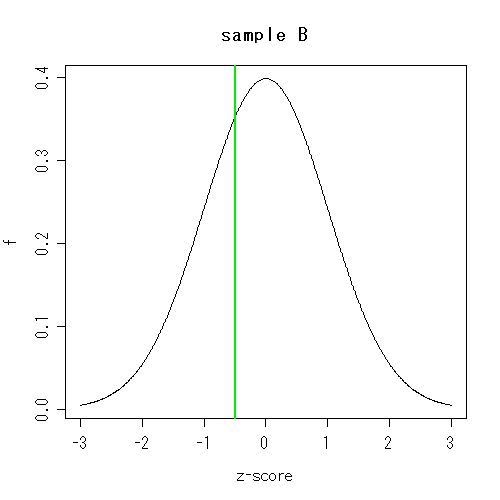 |
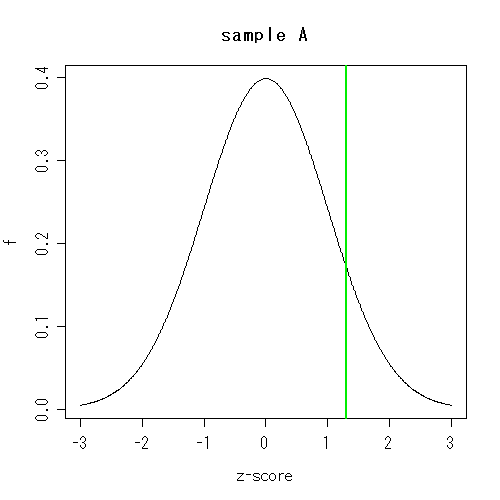
これらのサンプルは両方とも、遺伝子発現のレベル(の対数値)が正規分布している。
標準化された遺伝子発現のレベルはz-scoreで表される。
このスコアは、平均が0で、標準偏差が1になる性質を持っている
(その分布を示しているのが実線で書かれたヒストグラムだ)。
同じ遺伝子のスコアはサンプル間で直接に比較できる。
ここで一つの仮定をしている。
これらのサンプルの母集団の性質が同じであるか、同じであると見なせる、という仮定だ。
これらのサンプルの母集団の性質が同じであるか、同じであると見なせる、という仮定だ。
母集団の性質は同じか?
どちらも(測定の目的にもよるが、おそらく同じ種類の細胞の)mRNAの集団である。
これらは細胞を形成する基盤であるから、
本質的に異なることはたぶんないだろう。
おそらく分布中心は同じモル濃度になるだろう。
また分布の幅も同じだろう(こちらは多くの測定結果から支持されている)。(-> たとえば、この論文の例)
分布中心を実際に知るのは現時点では困難だ。
しかし仮に、分布中心のモル濃度が異なっていたとする---
細胞の種類が違うのならこれは有り得ることかもしれない*。
こうした場合、データを比較する目的は何になるだろう?
おそらくその目的は、
「ある遺伝子が、それぞれの細胞にとって、どんな役割を果たしているかを知ること」
「その役割がサンプル間で異なっているかどうかを知ること」
であろう。
トランスクリプトームはプロテオームを決定する。
そこで、ある遺伝子の役割は、その発現量が他の遺伝子の発現量という母集団のなかで、
どんな位置にあるのかによって決まるだろう。
これが妥当な推定であるなら、それぞれの母集団の性質は実質的に同等だと考えて良いことになる。
*タンパクの新陳代謝が低い組織なら、mRNAのモル濃度は低くてもかまわない。
プロテオームを保ちながら新陳代謝を下げるためには、タンパクの分解速度と合成速度とが等しく下がればよい---
たとえば、ある状態からそれぞれ半減させれば、プロテオームを保つことができる。
mRNAはタンパク合成のための鋳型であり、おそらく合成速度はこのモル濃度に比例する。
そこで、全てのタンパクの合成速度を半減させるためのもっとも単純な方法は、
mRNAのモル濃度を半分にすることだ。
新陳代謝を低くすれば、急激な変化に迅速に対応できないものの、省エネルギーな運用が可能だ。
ただ、細胞が実際にこのように合目的にできているかどうかはわからない。
プロテオームを保ちながら新陳代謝を下げるためには、タンパクの分解速度と合成速度とが等しく下がればよい---
たとえば、ある状態からそれぞれ半減させれば、プロテオームを保つことができる。
mRNAはタンパク合成のための鋳型であり、おそらく合成速度はこのモル濃度に比例する。
そこで、全てのタンパクの合成速度を半減させるためのもっとも単純な方法は、
mRNAのモル濃度を半分にすることだ。
新陳代謝を低くすれば、急激な変化に迅速に対応できないものの、省エネルギーな運用が可能だ。
ただ、細胞が実際にこのように合目的にできているかどうかはわからない。
細胞に含まれるRNA量は一定か?
上記に似た設問だが、この答えは否である。
対数正規分布する数列の和は不安定だからだ。
たぶん、もっとも強い発現を示す数種類の遺伝子が発現を変えれば
全RNA量は影響される。
これは、全RNA量でデータを標準化できないことの(原理的な)理由である。
一定な性質が期待されるのは分布の中心であり、それは
物理的な存在ではない。
対数正規分布する数列の和は不安定だからだ。
たぶん、もっとも強い発現を示す数種類の遺伝子が発現を変えれば
全RNA量は影響される。
これは、全RNA量でデータを標準化できないことの(原理的な)理由である。
一定な性質が期待されるのは分布の中心であり、それは
物理的な存在ではない。
同じサンプル内の、異なる遺伝子のz-scoreは比較できるか?
できるが、感度補正が必要になる。
現時点ではz-scoreは(モル濃度+感度)を表している(-> 論文Konishi 2008a)。
この感度はスポットとウエット実験の条件によって決まる定数である。
発現量の違い=(変化後モル濃度+感度spot1)- (変化前モル濃度+感度spot2)
そこで、同じスポットでの発現量の違いを求める際には、感度は相殺される。
しかし異なる感度をもつスポット間ではいつも感度spot1-感度spot2という決まった違いを生じる。
現時点ではz-scoreは(モル濃度+感度)を表している(-> 論文Konishi 2008a)。
この感度はスポットとウエット実験の条件によって決まる定数である。
発現量の違い=(変化後モル濃度+感度spot1)- (変化前モル濃度+感度spot2)
そこで、同じスポットでの発現量の違いを求める際には、感度は相殺される。
しかし異なる感度をもつスポット間ではいつも感度spot1-感度spot2という決まった違いを生じる。