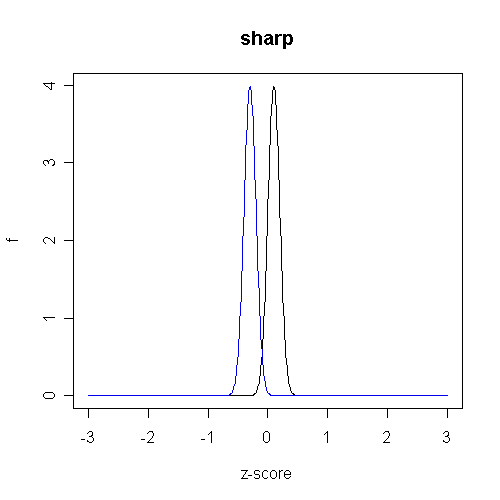
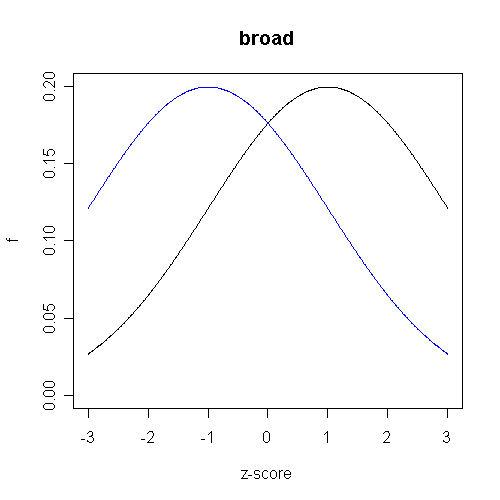
マイクロアレイは、分子生物学の測定方法のなかでは際立って項目数(スポット数)が多い。
そのため、偽陽性False Positiveの数も多くなる。これは困ったことだ。
偽陽性を根絶することは不可能だけど、
自分たちが耐えられるだけの数にコントロールすることはできる。
これはパラメトリック統計の得意分野のひとつである。
ここではそのための方法を概説する。
ときどき、多重検定の問題とごっちゃになっている解説を見かけるのだけど、
マイクロアレイで偽陽性が増えるのは、項目数が多いからである。
もし組織Aと組織Bの間に差があるかどうかが問題になっていて、
それを2組のマイクロアレイデータから検定しようとしているのなら、
これは(スポット数ぶんの)多重検定である。
けれど、これは設問がそもそもナンセンスだ。
どのトランスクリプトームも、少しずつ違っているものだから。
調べている組織が増えていって、たとえば組織Aから組織Gまでの間で
一度でも変動したことがある遺伝子を見つける、
なんていう際には多重検定の問題が生じる。
まあしかしこれはよくあるケースであって、マイクロアレイが特別なわけではない。
(そして、この種の試みをする場合には、偽陽性は深刻な問題にならないことが多い。
これはいわゆる宝探しであって、有望な候補は必ず追試されることになる)。
マイクロアレイで偽陽性が増えるのは、項目数が多いからである。
もし組織Aと組織Bの間に差があるかどうかが問題になっていて、
それを2組のマイクロアレイデータから検定しようとしているのなら、
これは(スポット数ぶんの)多重検定である。
けれど、これは設問がそもそもナンセンスだ。
どのトランスクリプトームも、少しずつ違っているものだから。
調べている組織が増えていって、たとえば組織Aから組織Gまでの間で
一度でも変動したことがある遺伝子を見つける、
なんていう際には多重検定の問題が生じる。
まあしかしこれはよくあるケースであって、マイクロアレイが特別なわけではない。
(そして、この種の試みをする場合には、偽陽性は深刻な問題にならないことが多い。
これはいわゆる宝探しであって、有望な候補は必ず追試されることになる)。
一方、偽陰性をコントロールすることはできない。
それは何か別の数学的なフレームワークを必要とするだろう。
帰無仮説が棄却できないときは黙るしかない。
差はなかったのだとは言えない。
それは何か別の数学的なフレームワークを必要とするだろう。
帰無仮説が棄却できないときは黙るしかない。
差はなかったのだとは言えない。