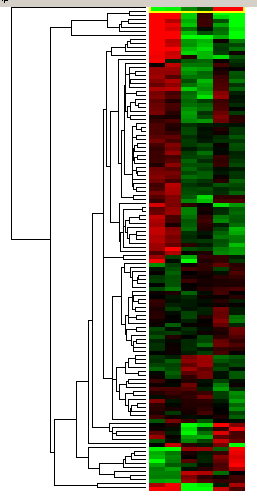
遺伝子群や、実験間の関係を考えるときに、ちょっとその関係を視覚化できると考える助けになるだろう。
クラスタリングはこのための方法である。
考え方として、「似たものを集める」方法と、「集団を区切る」方法がある。
階層型クラスタリングは前者であり、似ているものをグルーピングしたデンドログラムを出力する。→
測定値のzスコア、またはその平均からの差とって、
そのひとつひとつの中から最も近いもの同士をクラスターにして平均をとり、
その平均と最も近い次のクラスターとをまとめて平均をとり、...
そして最終的にひとつのクラスターにまとめるまで計算をする。
注意すべきアーティファクトとして、chaining という現象がある。
平均をとりつづけると「とんがった特徴」がなくなっていくので、
そのクラスターが「どれからも近い」無敵状態になってしまう。
これは、大きな平均クラスターが、
雪だるま式に小さいクラスターをまきこんでいくことから発見できる。
また、たぶんこれと関連するのだけど、
本来は似ているのに(統合の順番の問題で)仲間外れになってしまうこともある。
右のケースでも、たとえば一番下の2データのクラスターは
ちょっとワリを喰っている感がある。
「集団を区切る」方法であり、k-means法や自己組織化写像SOMなどの方法がある。
De Hoonさん作のclusterというソフトウエアでも実行される。
実験間の関係を視覚化する、という目的ではあまり使わないかもしれない。